

Todos os seres vivos são codificados por um conjunto de bases nitrogenadas: A (adenina), C (citosina), T (timina) e G (guanina). Dependendo da dedicação do leitor (a) à matéria de biologia na escola, será fácil lembrar que essas são as unidades formadoras do DNA, também conhecido como código da vida. A grosso modo, podemos dizer que o DNA é chamado de código da vida pois é uma espécie de repositório de informações, utilizado para implementar as funções biológicas de um organismo vivo.
Diga-se de passagem, é um repositório descentralizado, tendo em vista que, para organismos multicelulares, cada célula tem sua própria cópia do DNA. Para conseguir implementar as mais diversas funções biológicas, o DNA precisa passar pelos processos de transcrição e tradução, e esses mecanismos fazem parte do dogma central da biologia molecular.
De forma bastante simplificada, podemos resumir os processos de transcrição e tradução da seguinte forma: inicialmente, ocorre o processo de transcrição, no qual o DNA é convertido em RNA. Para os “computeiros” – como são chamados os profissionais de computação quando estão no grupo de biólogos -, o processo de transcrição pode ser entendido como uma espécie de compilação do código-fonte (DNA) em código objeto (RNA).
Na sequência, ocorre o processo de tradução, em que cada três bases de RNA (códon) serão utilizadas para codificar um aminoácido. No fim do processo, teremos uma cadeia de aminoácidos que formará uma proteína. Se quisermos dar seguimento à analogia anterior, o processo de tradução poderia ser entendido como a transformação do código objeto (RNA) em executável (proteína).
Dentro de um sistema biológico, a grande maioria das funções é executada por proteínas. Entretanto, o código-fonte que produz cada proteína é o DNA e, assim como em um programa de computador, podemos olhar o código-fonte para tentar entender como as funções estão sendo produzidas. Portanto, se quisermos entender como a vida funciona, precisamos inicialmente ler o código.Nesse caso, o código de DNA.
Em um segundo momento, temos que interpretar a função de cada trecho do código e como essa função afeta o sistema como um todo. Por fim, a missão mais complicada, alterar o código e, por consequência, a função.
Sequenciamento: a leitura do código da vida
O processo de sequenciamento consiste em ler as bases (A, C, T, G) formadoras do DNA de uma amostra. O primeiro método de sequenciamento foi criado por Frederick Sanger, ainda na década de 70, e era totalmente manual.
Embora o método de Sanger tenha sido mecanizado algum tempo depois, sequenciar genomas por esse método era muito demorado e caro. Com o surgimento dos sequenciadores de nova geração (NGS), houve uma explosão no número de sequências geradas. Além disso, o custo de sequenciamento vem caindo em um ritmo maior que a famosa lei de Moore.
Atualmente, estamos gerando mais dados que a nossa capacidade de analisar. Na tentativa de ajudar a resolver esse problema, há um grande esforço na evolução de algoritmos, heurísticas e ferramentas empregadas na análise desses dados.
Interpretando o código
Na bioinformática, é comum a utilização de diversos algoritmos e heurísticas na busca de significados biológicos das sequências de DNA e proteínas. Certamente, um dos algoritmos mais utilizados é o de programação dinâmica.
Diferentes tipos de programas, tais como buscadores de sequências (BLAST), programas para alinhamento múltiplo de sequências (CLUSTALW), buscadores de perfil (HMMER), programas buscadores de gene (GENSCAN) e programas para inferência filogenética (PHYLIP), além de outros, também utilizam o algoritmo de programação dinâmica ou uma aproximação que seja mais rápida. Portanto, embora existam diversos algoritmos na bioinformática, neste artigo, abordaremos como o algoritmo de programação dinâmica é empregado na busca por similaridade entre sequências de DNA ou proteínas.
Aviso aos navegantes: não espere código de implementação nem grande formalismo matemático, já que abordaremos os conceitos da programação dinâmica de uma maneira mais leve. Além disso, a implementação desse algoritmo em diferentes linguagens pode ser facilmente encontrada em uma busca rápida na Internet.
Alinhamento de sequências com algoritmo de programação dinâmica
Quando temos duas sequências de DNA ou proteínas, é comum o desejo de saber se elas são descendentes do mesmo organismo e/ou se têm as mesmas funções biológicas. Para ajudar nessa tarefa, nós vamos calcular um score que reflita o quanto essas sequências são similares.
É importante dizer que as sequências podem ser diferentes, não apenas por um processo de substituição simples (ex. A por T), mas também podem ocorrer processos de inserção e deleção em ambas as sequências e, portanto, uma base qualquer pode simplesmente sumir ou ser adicionada na sequência. Dessa forma, nós queremos otimizar o alinhamento dessas duas sequências para maximizar a similaridade.
Então parece simples, porque basta calcular o score de alinhamento para todas as possibilidades de alinhamento e escolher o melhor, certo? Sim, certo. A questão é que calcular todas as possibilidades de alinhamento é impraticável, pois existem por volta de 22N/2√πN diferentes alinhamentos para duas sequências de tamanho N.
Para se ter uma ideia, existem por volta de 10^179 alinhamentos possíveis entre duas sequências com 300 bases. Portanto, precisamos de um algoritmo mais sofisticado para encontrar alinhamentos ótimos – esses alinhamentos são aqueles de pontuação (score) máxima – e, nesse caso, utilizaremos programação dinâmica para encontrá-los.
O algoritmo de programação dinâmica utiliza o paradigma da divisão e conquista. Nesse paradigma, o objetivo é quebrar o problema em subproblemas que são similares ao problema original – recursivamente resolvem-se os subproblemas, e finalmente combinam-se as soluções para resolver o problema original.
O paradigma de divisão e conquista resolve subproblemas recursivamente, onde cada subproblema deve ser menor que o problema original. A seguir, demonstraremos a utilização da programação dinâmica para encontrar alinhamentos ótimos entre duas sequências.
Vamos definir duas sequências, X e Y. Inicialmente, é preciso encontrar as soluções dos subproblemas e armazená-las em uma tabela (matriz). No cenário do alinhamento de duas sequências X e Y, um subproblema é qualquer alinhamento entre X’ e Y’ tal que: X’ = um prefixo de X e Y’ = um prefixo de Y. Então, para encontrar a solução ótima, deve-se ir achando as soluções dos subproblemas menores para os maiores.
O último elemento da tabela a ser preenchido conterá a solução “completa” do problema. Não se preocupe em entender tudo nesse momento, isso deve ficar um pouco mais claro quando analisarmos o preenchimento da matriz de programação dinâmica.
Tipos de alinhamentos
Considere que, para as sequências X=TTCATA e Y=TGCTCGTA, existem três possibilidades de alinhamento entre as bases da sequência X e Y.
- x alinhado com y – match (igual) ou mismatch (desigual).
- x alinhado com gap – O gap (espaço) representa os eventos de inserção ou deleção nas sequências.
- y alinhado com gap.
Sistema de pontuação
Também precisamos definir uma pontuação para cada alinhamento. Quando x for alinhando com y, se x=y (match), então o valor do alinhamento será 5. Caso contrário, se x≠y (mismatch), o valor do alinhamento será -2.
Quando x=gap ou y=gap, o alinhamento será penalizado com o valor de -6. Esse sistema de pontuação pode variar, e os softwares que implementam esse algoritmo costumam informar os valores do seu sistema de pontuação.
Inicializando a matriz de programação dinâmica
Agora que temos as possibilidades de alinhamentos e um sistema de pontuação, podemos inicializar a matriz de programação dinâmica, figura 1. Primeiro, devemos alinhar a sequência X (x1..xm) e Y (y1..yn) com uma cadeia vazia.
Esse alinhamento está representado em nossa matriz pela coluna 1 (esquerda) e linha 1 (superior), respectivamente. Além disso, a primeira célula da matriz (superior esquerda) está alinhando x0..y0, ou seja, nada com nada e, portanto, deve ser inicializada com o valor 0 (zero).
Definição do algoritmo recursivo
Vamos escrever uma definição recursiva geral para todos os nossos resultados (escores) de alinhamento ótimo parcial S(i,j):
- S(i,j) = max [S(i-1, j-1) + matriz(i,j),
- S(i-1, j) + penalidadeDoGap(),
- S(i, j-1) + penalidadeDoGap()]
- se i=j, então matriz(i,j) = 5
- se i!=j, então matriz(i,j) = -2
- penalidadeDoGap() = -6
Com a matriz inicializada, ou seja, a linha superior e a coluna esquerda preenchidas, é possível completar o resto da matriz usando a definição recursiva S(i,j). Dessa maneira, podemos calcular o valor de qualquer célula para a qual saibamos os valores das três células adjacentes, para a parte superior esquerda (i-1, j-1), acima (i-1, j) e para a esquerda (i, j-1).
Existem várias maneiras diferentes de fazer isso: uma delas é iterar dois loops aninhados, i = 1 para M e j = 1 para N. Assim, estaremos preenchendo a matriz da esquerda para a direita, de cima para baixo.
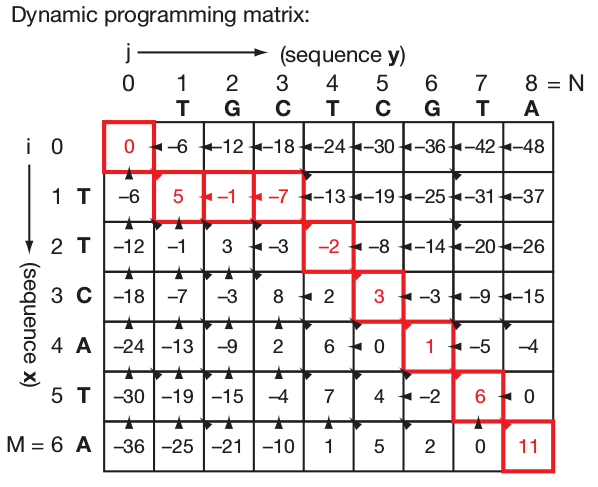
Enfim, o alinhamento ótimo
Uma vez que terminamos de preencher a matriz, a pontuação do alinhamento ótimo das sequências completas é a última pontuação que calculamos. De qualquer forma, ainda não conhecemos o alinhamento ótimo. Para descobri-lo, podemos fazer um rastreamento (traceback) recursivo na matriz.
Começamos na célula (m, n), para qual temos que determinar quais dos três casos (superior esquerda (i-1, j-1), acima (i-1, j) ou para a esquerda (i, j-1)) usamos para chegar aqui – por exemplo, podemos repetir os mesmos três cálculos e registrar essa escolha como parte do alinhamento e, em seguida, seguir o caminho apropriado; nesse caso, de volta à célula anterior no caminho ideal.
Temos que fazer isso em uma célula no caminho ótimo por vez, até atingirmos a célula (0,0), altura em que o alinhamento ótimo é totalmente reconstruído. Podemos observar o traceback do caminho ótimo do nosso alinhamento em vermelho e com as setas indicativas na figura 1. O alinhamento ótimo pode ser visto na figura 2.

Considerações finais
O algoritmo de programação dinâmica não é trivial e seu tempo de execução para duas sequências é quadrático. Em outras palavras, para sequências suficientemente grandes, ele também pode ser impraticável. Entretanto, esse algoritmo garante matematicamente o melhor alinhamento (de pontuação mais alta).
Embora isso não signifique que o alinhamento ótimo corresponda a um alinhamento biologicamente correto, esse é um problema para o sistema de pontuação, não para o algoritmo.
De forma similar, o algoritmo de programação dinâmica é capaz de alinhar com sucesso sequências não relacionadas. Por exemplo, as duas sequências aqui apresentadas, X e Y, podem parecer bem alinhadas, contudo elas não são relacionadas. São sequências de DNA geradas aleatoriamente! A questão de quando uma pontuação é estatisticamente significativa também é um problema separado, que requer uma dose considerável de estatística.
Referências
- J.C. Setubal Chapter A05 Similarity Search – https://goo.gl/q3ow3v
- Eddy, Sean R. “What is dynamic programming?.” Nature biotechnology 22.7 (2004): 909-910.
Site com implementação do algoritmo de programação dinâmica: https://goo.gl/5YfWta.
A bioinformática é uma área multidisciplinar formada principalmente por biologia, computação e estatística. O Instituto de Matemática e Estatística da USP oferece cursos de mestrado e doutorado em bioinformática. https://www.ime.usp.br/bioinfo/pos.
***
Artigo publicado na revista iMasters, edição #24: https://issuu.com/imasters/docs/24

De 0 a 10, o quanto você recomendaria este artigo para um amigo?